8 Minuten
Zellen sind auf Mitochondrien als Energiequellen angewiesen; neue Forschungsergebnisse verbinden jedoch Fehler bei der Replikation der mitochondrialen DNA (mtDNA) mit chronischen Entzündungsreaktionen, die im Alter auftreten. Wissenschaftlerinnen und Wissenschaftler des Max-Planck-Instituts für Biologie des Alterns und Kooperationspartner untersuchten menschliches Gewebe sowie altersassoziierte Mausmodelle und identifizierten einen molekularen Mechanismus: Mitochondrien stoßen Fragmente ihrer eigenen DNA aus der Organelle ins Zytoplasma aus, was dort entzündliche Signalwege aktiviert.
Wissenschaftlicher Hintergrund: mtDNA, Nucleotide und Zellalterung
Mitochondrien besitzen ein eigenes Genom – die mtDNA –, das bei der Erneuerung und Teilung der Organellen stets fehlerfrei kopiert werden muss, um die Funktion zu erhalten. Für die Replikation von Nukleinsäuren nutzt der Zellkern Deoxyribonukleotide (dNTPs) als Bausteine. Zahlreiche frühere Studien zeigen, dass die Verfügbarkeit dieser dNTP-Pools in gealterten oder seneszenten Zellen abnimmt, was die DNA-Synthese generell gefährden kann und Reparaturprozesse belastet.
Verringern sich die dNTP-Vorräte, können mitochondriale Polymerasen als Folge versehentlich Ribonukleotide (rNTPs) in die DNA einbauen. Solche Ribonukleotide destabilisieren lokal die DNA-Struktur, führen zu fehlerhaften Kopien oder Lücken und erzeugen Lesionen, die das Organell als beschädigt erkennt. In der Folge kann die Mitochondrienmembran Partien der fehlerhaften mtDNA ins Zytosol entlassen. Zytosolische mtDNA wird von angeborenen Immunrezeptoren erkannt und löst entzündliche Signalwege aus, die langfristig zu Gewebeschäden beitragen können.
Studiendesign und zentrale Ergebnisse
Die Forschenden kombinierten Analysen an menschlichen Gewebeproben mit genetisch veränderten Mausmodellen, die altersähnliche Abfälle in Stoffwechsel- und Reparaturwegen nachbilden. Mithilfe biochemischer Tests, hochauflösender Sequenzierung und histologischer Techniken verfolgte das Team, wie eine veränderte Balance der Nukleotid-Pools die Integrität der mtDNA beeinflusst und welche Folgen dies für zelluläre Entzündungsprozesse hat. Die Untersuchungen zeigten, dass in gealterten oder dNTP-armen Geweben die Einbaurate von Ribonukleotiden in die mtDNA deutlich anstieg. Diese unvollkommenen mtDNA-Moleküle waren verstärkt dazu geneigt, aus den Mitochondrien ausgestoßen zu werden.
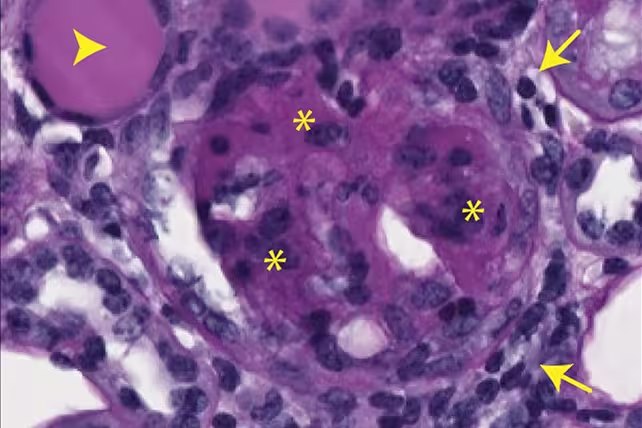
Hinweise auf Nierenvernarbungen bei Mäusen, verursacht durch ausgelagerte mtDNA. (Max Planck Institute for Biology of Ageing)
Die ins Zytoplasma gelangte mtDNA aktivierte Immunwege, die mit chronischer Entzündung und Gewebenarbenbildung verknüpft sind. In den Tiermodellen korrelierte die Menge an zytosolischer mtDNA mit Markern entzündlicher Aktivierung und mit histologisch nachweisbarer Fibrose. Die Studie stellt damit eine direkte Verbindung zwischen einem metabolischen Ungleichgewicht – konkret dem Absinken der dNTP-Verfügbarkeit – und einer molekularen Kaskade her, die mitochondriale Dysfunktion in entzündliche Alterungsphänotypen überführt.
Wichtige Aussagen der Forschenden
Thomas Langer vom Max-Planck-Institut für Biologie des Alterns fasst die Schlussfolgerung so zusammen: Stoffwechselstörungen, die die Zusammensetzung der Nukleotid-Pools verändern, können eine Kaskade in Gang setzen, die in seneszenten Zellen und gealterten Organen entzündliche Prozesse auslöst und damit potenzielle Ansatzpunkte für Interventionen bieten. Dusanka Milenkovic, ebenfalls am Institut tätig, weist darauf hin, dass bereits heute eingesetzte Therapien für bestimmte mitochondriale Erkrankungen – etwa die Gabe von DNA-Vorläufern oder Nukleosiden – geprüft werden sollten, ob sie auch altersassoziierte Entzündungen abschwächen können. Diese Idee stützt sich auf die Vorstellung, dass eine Stabilisierung der dNTP-Pools die Fehlincorporation von rNTPs reduzieren könnte.
Auswirkungen auf Gesundheit und Perspektiven für künftige Forschung
Dieser Mechanismus liefert eine plausible Erklärung dafür, wie sich über Jahrzehnte hinweg niedriggradige, chronische Entzündungen ansammeln und zur Entstehung häufiger altersassoziierter Erkrankungen beitragen können – dazu zählen bestimmte Krebsarten, kardiovaskuläre Erkrankungen und neurodegenerative Leiden wie die Alzheimer-Krankheit. Wenn es gelingt, die Fehleinlagerung von Ribonukleotiden zu verhindern oder die Stabilität der mtDNA so zu erhöhen, dass sie nicht aus den Mitochondrien ausgestoßen wird, könnte dies die schädliche entzündliche Signalgebung in älterem Gewebe reduzieren.
Als konkrete Forschungsrichtungen schlagen die Autoren mehrere Ansatzpunkte vor: erstens, die quantitativen Analysen darüber zu vertiefen, wie häufig dieser Weg im normalen menschlichen Altern gegenüber spezifischen Krankheitszuständen aktiv wird; zweitens, Sicherheits- und Wirksamkeitsstudien zu entwickeln, die untersuchen, ob eine pharmakologische oder diätetische Wiederherstellung der dNTP-Pools Entzündungsmarker und Gewebeschäden verringert; und drittens, die molekularen Erkennungswege für zytosolische mtDNA weiter zu entschlüsseln, um gezielte Blockaden zu testen.
Technisch könnten in nachfolgenden Studien detaillierte Massenspektrometrie-Analysen der Nukleotid-Pools (LC-MS), spezialisierte Sequenzierungsverfahren zur Erkennung eingebauter rNMPs in mtDNA sowie Live-Imaging und Elektronenmikroskopie zur Visualisierung von mtDNA-Exporten eingesetzt werden. Ebenso relevant sind genetische Experimente, die Enzyme modulieren, welche an der dNTP-Synthese beteiligt sind – etwa Ribonukleotidreduktase (RNR) oder die mitochondrialen Nukleotidkinasen wie TK2 – um kausale Zusammenhänge streng zu prüfen.
Gleichzeitig müssen Risiken einer therapeutischen dNTP-Erhöhung untersucht werden: Eine unspezifische Anhebung der dNTP-Spiegel könnte zelluläre Mutationsraten verändern oder proliferative Zellen fördern, was potenziell tumorigen wirken kann. Daher sind sorgfältig dosierte, tissuespezifische Ansätze sowie biomarkerbasierte Stratifikationen von Patientengruppen zentral, bevor klinische Studien geplant werden.
Technische Details und molekulare Mechanismen
Auf molekularer Ebene beruht das beobachtete Phänomen auf mehreren miteinander verzahnten Mechanismen. Zunächst verschiebt ein Mangel an dNTPs das Substratverhältnis zugunsten von rNTPs. Polymerase gamma, die für die mtDNA-Replikation verantwortlich ist, besitzt zwar Mechanismen zur Vermeidung von Fehlpaarungen, kann aber bei hoher rNTP-Verfügbarkeit und niedrigem dNTP-Spiegel dennoch Ribonukleotide einbauen. Eingebaute rNMPs schwächen die Helix und machen die DNA anfälliger für Abspaltungen, Konformationsänderungen und Reparaturintermediäre. Solche strukturellen Störungen können die Mitochondrien-Qualitätskontrolle, inklusive Mitophagie und Membran-Remodellierung, aktivieren.
Ein weiterer Befund betrifft die zellulären Sensoren, die zytosolische DNA erkennen: Das cGAS-STING-System ist ein zentraler Weg, mit dem zelluläre Abwehr-Reaktionen gegen cytosolische dsDNA ausgelöst werden und Interferon- und NF-κB-abhängige Transkriptionsprogramme ankurbeln. Zusätzlich können Inflammasom-Komponenten wie NLRP3 und endosomale Rezeptoren wie TLR9 auf mtDNA reagieren — jeweils mit leicht unterschiedlichen Konsequenzen für die Zytokinfreisetzung und das Gewebeverhalten. Die Studie deutet darauf hin, dass ausgelagerte mtDNA mehrere dieser Pfade aktivieren kann, was die anhaltende, niedriggradige Entzündungsantwort erklärt.
Schließlich spielt der zelluläre Nukleotidstoffwechsel eine Rolle: Die Balance zwischen synaptischen und de novo gebildeten dNTPs, die Aktivität von Nukleotid-Transportern und Mitochondrien-spezifischen Kinasen sowie Katabolismuswege beeinflussen die Verfügbarkeit der Bausteine. Alterungsbedingte Veränderungen in diesen Systemen – etwa verringerte Expression von Enzymen der dNTP-Synthese oder erhöhte Aktivität von dNTP-hydrolisierenden Proteinen – können die Prädisposition für rNMP-Einbau erhöhen.
Translationale Bedeutung und potenzielle Interventionen
Die Übersetzung der Ergebnisse in klinische Anwendungen erfordert ein mehrstufiges Vorgehen. Kurzfristig könnten präklinische Studien testen, ob die Ergänzung mit Nukleosiden oder deren Vorstufen in Tiermodellen die mtDNA-Integrität verbessert, die Menge zytosolischer mtDNA reduziert und damit auch Entzündungsmarker senkt. Mittel- bis langfristig wären humanbasierte Pilotstudien denkbar, die auf älteren Probandengruppen mit Biomarker-getriebener Selektion basieren. Bereits zugelassene Therapien für seltene mitochondriale Syndromzustände, welche Nukleosid-Substitutionen nutzen, bieten einen rationalen Ausgangspunkt für Wirkungs- und Sicherheitsstudien in einem Alterskontext.
Parallel lässt sich an zielgerichteten Ansätzen arbeiten: Kleiner Moleküle, die die Fehlerrate von Polymerase gamma senken; Inhibitoren oder Modulatoren von DNA-Erkennungswegen (z. B. cGAS- oder STING-Inhibitoren) zur Reduktion pathologischer Immunantworten; oder Strategien zur gezielten Verstärkung mitochondrialer Qualitätskontrolle wie selektive Mitophagie. Jede dieser Optionen bringt spezifische Chancen und Risiken mit sich und bedarf detaillierter präklinischer Evaluierung.
Wissenschaftlich besonders wertvoll ist die mögliche Identifizierung von Biomarkern, die anzeigen, wann dieser Mechanismus in Patienten relevant ist: Messungen der rNMP-Last in mtDNA, Profile von dNTP-Pools in Gewebeflüssigkeiten, sowie entzündliche Zytokinsignaturen könnten helfen, Patienten zu stratifizieren und Therapieansprechen besser zu prognostizieren.
Fazit
Die Studie verbindet einen metabolischen Engpass – die Reduktion von deoxyribonukleotiden – mit einem klaren molekularen Weg hin zu Entzündungsreaktionen über mtDNA-Instabilität und deren Ausstoß aus Mitochondrien. Diese Erkenntnisse eröffnen gezielte Ansatzpunkte, um die Integrität des mitochondrialen Genoms zu erhalten und möglicherweise entwicklungstypische, inflammation-getriebene Funktionsverluste im Alter abzubremsen. Entscheidend werden umfangreiche präklinische Untersuchungen und sorgfältig konzipierte klinische Studien sein, um zu prüfen, ob Nukleotid-Supplementierung, Modulation der Polymeraseaktivität oder Hemmung von DNA-Erkennungspfade sicher in messbare gesundheitliche Vorteile für ältere Menschen übersetzt werden können.
Quelle: sciencealert
Kommentar hinterlassen